生信常见文件的格式以及查看与处理方法
生信分析过程中,会与很多不同格式的文件打交道,除了原始测序数据fastq之外,还需要准备基因组文件fasta格式和基因注释文件gtf格式。在分析的过程中还会有众多中间文件的生成,如bed、bed12、sam、bam、wig、bigwig、bedgraph、vcf等,生成后我们一般会查看下内容了解文件每一列的含义,以此来决定需要提取哪些有用信息列来进行下一步分析。
一、测序数据FASTQ文件
1)文件用途:样品测序返回的数据一般存储为fastq文件,通常是压缩文件filename.fq.gz的格式,节省存储空间和传输时间。NGS基础 - FASTQ格式解释和质量评估
2)查看方式
1 | # zcat查看gzip压缩的文件 |
3)格式说明:fastq文件每4行代表一条序列 第一行:记录序列测序时所用仪器以及在测序通道中坐标信息,以@开头; 第二行:测序的序列信息,以ATCGN表示,由于荧光信号干扰无法判断是什么碱基时就用N表示; 第三行:通常一个+; 第四行:与第二行碱基信息一一对应,存储测序碱基的质量值。
4)其他常用命令
1 | # 计算read数 |
awk的介绍见:常用和不太常用的awk命令
二、基因组FASTA文件
此文件可以从ensemble数据库下载的(https://www.ensembl.org/info/data/ftp/index.html), 一般选择下载primary assemblyfasta(想知道为什么,点这里)。fasta文件用于序列存储,可以是DNA或蛋白序列,在此FASTA文件存储了基因组序列的信息。
序列名字行:以>符号开头,记录了该序列类型和所在基因组位置信息;
序列行(一行或多行):序列信息,soft-masked基因组会把所有重复区和低复杂区的序列用小写字母标出的基因组,小写字母n表示未知碱基。
1 | >1 dna_sm:chromosomechromosome:GRCh38:1:1:248956422:1 REF |
三、基因组注释文件gff和gtf
gff全称General featureformat,主要是用来注释基因组。gtf全称Gene transfer format,主要是用来对基因进行注释。两者均是一个9列的基因信息注释文件,前8列的信息几乎一样,区别在于第9列。具体可见历史推文NGS基础 - NGS基础 - GTF/GFF文件格式解读和转换
GFF文件是以tab键分割的9列组成,以下为每一列的对应信息:
- seq_id:序列的编号,一般为chr或者scanfold编号;
- source: 注释的来源,一般为数据库或者注释的机构,如果未知,则用点“.”代替
- type: 注释信息的类型,比如Gene、cDNA、mRNA、CDS等;
- start: 该基因或转录本在参考序列上的起始位置;(从1开始,包含);
- end: 该基因或转录本在参考序列上的终止位置;(从1开始,包含);
- score: 得分,数字,是注释信息可能性的说明,可以是序列相似性比对时的E-values值或者基因预测是的P-values值,
.表示为空; - strand: 该基因或转录本位于参考序列的正链(+)或负链(-)上;
- phase: 仅对注释类型为“CDS”有效,表示起始编码的位置,有效值为0、12. (对于编码蛋白质的CDS来说,本列指定下一个密码子开始的位置。每3个核苷酸翻译一个氨基酸,从0开始,CDS的起始位置,除以3,余数就是这个值,,表示到达下一个密码子需要跳过的碱基个数。该编码区第一个密码子的位置,取值0,1,2。0表示该编码框的第一个密码子第一个碱基位于其5’末端;1表示该编码框的第一个密码子的第一个碱基位于该编码区外;2表示该编码框的第一个密码子的第一、二个碱基位于该编码区外;如果Feature为CDS时,必须指明具体值。);
- attributes: 一个包含众多属性的列表,格式为“标签=值”(tag=value),以多个键值对组成的注释信息描述,键与值之间用“=”,不同的键值用“;”隔开,一个键可以有多个值,不同值用“,”分割。注意如果描述中包括tab键以及“,= ;”,要用URL转义规则进行转义,如tab键用 代替。键是区分大小写的,以大写字母开头的键是预先定义好的,在后面可能被其他注释信息所调用。
从ensemble下载的gtf文件前5行一般是以#开头的注释信息,后续分析中用不上需要去除,同时需要给第一列添加chr标签(与基因组序列一致),可通过下面的命令对文件进行加工:
1 | # grep 匹配查询 -v 输出不匹配的行 |
四、bed文件
1、介绍
分析过程中的bed文件一般代表区域信息,如表示Peak位置的bed文件,表示基因注释的bed12文件。
表示基因注释时,gtf/gff和bed文件的区别:
1)gtf/gff文件一行表示一个exon/CDS等子区域,多行联合表示一个gene;bed文件一行表示一个gene; 2)gtf文件中碱基位置定位方式是1-based,而bed中碱基定位方式是0-based,如下图所示。

bed文件每一行对应信息
必须包含的3列信息: 1)chrom:染色体名字 (e.g.chr3, chrY, chr2_random或者scaffold10671)。 2)chromStart:基因在染色体或scaffold上的起始位置(0-based)。 3)chromEnd:基因在染色体或scaffold上的终止位置 (前闭后开)。
可选的9列信息: 4)name:bed文件的行名。 5)score:本条基因在注释数据集文件中的评分(0-1000),在Genome Browser中会根据不同区段的评分显示对应的阴影强度(评分越高灰度越高)。 6)strand:链的方向+、-或. (.表示不确定链的方向) 7)thickStart:CDS区(编码区)的起始位置,即起始密码子的位置。 8)thickEnd:The ending position at which the feature is drawn thickly (for example the stop codon in gene displays). 9)itemRgb:RGB颜色值(如:255,0,0),方便在GenomeBrowser中查看。 10)blockCount:bed行中外显子的数目。 11)blockSizes:逗号分割的列,数目与blockCount值对应,每个数表示对应外显子的碱基数。 12)blockStarts:逗号分割的列,数目与blockCount值对应,每个数表示对应外显子的起始位置(数值是相对ChromStart计算的)。
2、narrow, broad, gapped peak: 三种格式之间的区别与联系
在进行peak calling分析时,经常会接触到以下3种peak格式:narrow peaks format,broad peaks fotmat,gapped peaks format
peak被定义为基因组上一段reads富集的区域,核心信息是在染色体上的起始和终止位置,除此之外,还有软件对于该peak区域的打分,比如常见的pvalue, qvalue, fold_enrichment等值。
和基因组比对信息用BAM格式来存储类似,为了标准化不同peak calling软件的输出,特意制定了以上3种数据格式。这三种格式本质上都是bed文件,只不过列数不太类似。
(1)Narrow Peaks Format;
该格式又称之为point-source peaks format, macs2默认输出就是这种格式,是一种BED6+4的格式,列数为10列,如下:

前四列分别代表chrom, chromStart, chromEnd, name, 用于描述peak区间和名称,注意bed格式中起始位置从0开始计数。 第五列代表score,在macs2的输出结果中为int(-10*log10qvalue); 第六列代表strand, 在macs2的输出结果中为; 第七列代表signalvalue, 通常使用fold_enrichment的值; 第八列代表pvalue, 在macs2的输出结果中为-log10(pvalue); 第九列代表qvalue, 在macs2的输出结果中为-log10(qvalue); 第十列代表peak, 在macs2的输出结果中为peak的中心,即summit距离peak起始位置的距离。
(2)Broad Peaks Format
这种格式就是在narrow peaks format的基础上丢掉了最后一列的信息,为BED6+3的格式, 列数为9列。
(3)Gapped Peaks Format
前两种格式都是由于描述连续的peak区间,适用于DNA水平上的富集区域信息的存储,比如chip_seq, ATAC_seq鉴定到的peak区间,而gapped peaks format用于描述非连续的peak区间,这里的非连续通常指的是在peak的区间内会包含多个exon区域,适用于RNA水平上的富集区域信息的存储,比如m6A_seq鉴定到的peak区间。
该格式在BED12的基础上进行延伸,演变为BED12+3的格式,列数为15列,每列的含义示意如下

前6列的含义和上述两种peak格式完全相同,后3列的含义和broad peak完全相同,为了专区表示peak区间内包含的exon信息,借鉴转录本的BED12格式,引入了以下6列: thickStart,thickEnd,itemRgb,blockCount,blockSizes,blockStarts thickStart和thickEnd有点类似转录本中CDS的起始和终止位置,在存储peak信息时,通常的做法是将这两列的值和chromStart和chromEnd的值设置成相同的,itemRgb是一个RGB颜色值,比如255,0,0, 如果没有对应的颜色信息,则用0来表示。 blockCount代表该peak区间包含的exon的个数,blockSizes代表每个exon区间的长度,多个exon用逗号连接,blockStarts代表每个exon区间在基因组上的起始位置,多个exon用逗号连接。
关于这三种格式的相关介绍请参考以下链接
https://genome.ucsc.edu/FAQ/FAQformat.html#format13
3、bed数据处理
详细可以参考官方网页:The BEDTools Suite
中文参考文档:最全Bedtools使用说明--只看本文就够了
五、sam和bam文件
1、介绍
sam文件全称The SequencingAlignment/Map Format,是Alignment/Map步骤bwa/STAR/HISAT2等软件对结果的标准输出文件,用于存储reads比对到参考基因组的比对结果,是一个纯文本格式,文件一般较大。为了节省硬盘存储,一般使用其高效压缩的二进制格式bam文件。
利用samtools view的-b参数就能把sam文件转为bam文件。
1)sam文件查看方式 在linux终端直接用less即可进行查看;
2)bam文件查看方式 需要借助samtools view工具进行查看
1 | samtools view filename.bam | less -S |
NGS分析中大多数文件都是由header和record两部分组成,加上-h参数后可以将header显示出来,默认是不显示的。
1 | @HD VN:1.5 SO:coordinate |
header内容
1 | @HD:是必须的标准文件头,包含版本信息; |
record内容
1 | 每一行就是一条read比对上参考基因组的信息,总共12列,用tab键分割。 |
2、flag信息
sam文件中第二列flag信息很重要,下面做进一步解释。
利用samtools flagstat工具可以查看bam文件中比对的flag信息,并输出比对的统计结果。
samtools flagstat *.bam
flag一共有12个标签,使用16进制数表示,每个标签值是2^(n-1),其中n<=12,每个值有其对应的唯一解释含义,具体见下图。

1 | 1 : 代表这个序列采用的是PE双端测序 |
你会发现随机挑选几个值做加和运算,他们的结果都是唯一的,所以在bam文件中第二列flag的值代表这条序列符合下图所示条件的值的和。
所以根据这个值我们可以判断这条序列是双端测序还是单端测序;如果是双端测序,reads来自左端还是右端。比如65只能是1和64组成,代表这个序列是双端测序,而且是read1。
每次转换很头疼?别担心,网上有很多解码flag含义的在线工具,如SAM Format(网址:https://www.samformat.info/sam-format-flag)
输入flag的值,解析工具会返回匹配上的信息。如果是单端测序,flag值都是偶数。
如果是双端测序,工具会帮我们把另外一端序列的flag值返回,并且将这些数字情况大致分为5类,在右侧进一步显示这个值对应的含义。
3、sam/bam文件处理
详细可以参考官方网页:Samtools
中文参考文档:****samtools常用命令详解****
wig、bigwig和bedgraph文件
上述bam和sam文件可以帮助我们探索reads在参考基因组中的比对情况,导入基因组浏览器查看比对状态和突变信息。而wiggle(简称wig)、bigwig(简写bw)以及bedgraph(简写bdg)只包含区域和区域的覆盖度信息,文件更小,用于可视化更方便,可以导入基因组浏览器(Genome Browser)中进行可视化,以查看reads在参考基因组各个区域的覆盖度并检测测序深度。这几个文件在ChIP-seq数据分析Call Peak阶段会生成,可以利用IGV等工具进行可视化,方便查看组蛋白修饰的程度。
六、wig/bigwig介绍
wig 文件全称叫 Wiggle Track Format, 用来绘制基因组上的图形轨迹的文件格式。wig 格式是较老的格式,用来显示密集且连续的数据,比如GC含量,概率分数,转录组数据等,而bigwig是wig格式的压缩形式。
wig 数据有两种类型:variableStep 和 fixedStep。
我们先来研究这两种类型的文件,后面在基因组浏览器中利用这些文件显示轨迹。
1、variableStep 格式
(1)特点及适用场景:
- 在指定的染色体片段区域绘制条形图
- 用于全基因组数据集(大约百万分之十的数据点)
- 指定的区域必须为恒定大小(由span参数指定)
- 数据点间具有不规则间隔的数据,但是在某些情况下建议谨慎
- 如果数据点的不规则间距太极端,此格式在编码和显示期间可能效率很低。在这种情况下,“bedGraph”是最佳格式。
- 一般UCSC不建议采用该格式作为基因组浏览器输入文件,因为考虑到数据集大小与索引构建,都不如 bigwig 更高效
- 使用 wigToBigWig 将 Wig 转换为 bigWig 文件
(2)格式:
声明行:以单词variableStep开头,后跟染色体规范。
数据行:两列,分别包含染色体位置和数据值。
1 | variableStep chrom=chrN |
(3)例子:
下面表示在2号染色体上的300701-300705位置均显示12.5
1 | variableStep chrom=chr2 |
可选的span参数(默认值:span = 1)允许更简洁地指定由连续的具有相同数据值的碱基组成的数据。
跨度从指定的每个染色体位置开始,并指示数据值应覆盖的碱基数。例如: 应用span=5后,相当于:
1 | variableStep chrom=chr2 span=5 |
wiggle 格式用于快速显示非常密集的数据。当每1,024个碱基只有几个数据点时,variableStep格式会变得效率极低。 如果variableStep数据点相距大于约100个碱基,则建议使用 BedGraph格式。
2、fixedStep 格式
(1)特点及适用场景:
- 在指定的染色体片段区域绘制条形
- 最适合用于全基因组数据集(大约百万分之十的数据点)
- 指定的区域必须为恒定大小(由span参数指定)
- 染色体位置精确地有规律的间隔(由step参数指定)
(2)格式:
声明行:单词fixedStep开头,并包含染色体,起始坐标和步长的规范 。
数据行:一列,包含数据值。
1 | fixedStep chrom=chrN |
(3)例子:
表示3号染色体上的三个区域(每个区域5个碱基):400601-400605、400701-400705和400801-400805,分别显示值11、22和33
1 | fixedStep chrom=chr3 start=400601 step=100 span=5 |
对于variableStep和fixedStep格式,必须在整个数据集中使用相同的。如果未指定跨度,则使用默认跨度1。 顾名思义,fixedStep 在整个数据集中需要相同的大小步长。如果未指定,则步长为1。
3、数据值
wiggle 格式的数据值可以包含整数,实数,正值或负值,但不支持 NaN 值。未指定的位置没有数据,也就不会显示。
需要注意:wiggle文件(variableStep and fixedStep )的染色体坐标是从1开始。 例如,对于长度为N的染色体,第一个位置为1,最后一个位置为N。 对于 bigwig 文件,使用 wiggle 转换为 bigwig,使用的染色体坐标是从1开始。 而使用bedGraph格式创建的BigWig文件使用0开始。 自定义 wiggle 轨迹的参数:
1 | track type=wiggle_0 name=track_label |
其他参数:
1 | autoScale <on|off> # default is on |
4、例子
(1)下面是 Wig 格式的文件,分别包含 variableStep 和 fixedStep 格式创建的两段轨迹
1 | browser position chr19:49304200-49310700 |
(2) 选择菜单栏My Data的Custom Tracks

(3)将上面的代码粘贴到输入框,点 Submit

(4)简要信息显示,点Go进行绘制

(5)拿到绘制好的轨迹图

七、BedGraph(基因组浏览器绘制
1、特点及适用场景:
- 存放区间的坐标轴信息和相关评分(score)的文件,主要用于存储稀疏,不连续的数据
- 后缀名.bedGraph
- 一般UCSC不建议采用该格式作为基因组浏览器输入文件,因为考虑到数据集大小与索引构建,都不如 bigwig 更高效,尤其在如果bedGraph数据集非常大(超过5000万行 ),推荐转为 bigwig 文件
- 使用WigTobigWig将 bedGraph 转换为bigWig 文件
- 需要注意 bedGraph 文件不能转换为 wig 文件
2、格式
一共包含四列:chromA chromStartA chromEndA dataValue,有点类似bed文件 分别为:
chromA 染色体号 chromStartA 起始位点:染色体坐标从 0 开始,这意味着第一个染色体位置为0,而长度为N的染色体的最后位置将为N-1。 chromEndA 终止位点 dataValue 数据值:数据值可以是整数或实数,正值或负值。输入数据中列出的位置必须按数字顺序,并且仅会绘制指定位置的图。
参数:
1 | track type=bedGraph name=track_label description=center_label |
3、例子
下面的文件可以描述为:
在第19号染色体的49,302,001到49,304,701区域的三个轨迹中指定9个独立的数据点。
1 | browser position chr19:49302001-49304701 |
粘贴上面的代码,点 Submit
https://genome.ucsc.edu/cgi-bin/hgCustom

绘制轨迹

上面的示例是一个定制轨迹,其中包括track type = 一行特定于在浏览器中加载数据的行。 此行将导致原始bedGraph数据文件无法通过validateFiles浏览器外部的其他工具进行验证。
推荐大家阅读UCSC官网对这几个文件的详细解释:
wiggle(WIG):https://genome.ucsc.edu/goldenPath/help/wiggle.html
bedGraph:https://genome.ucsc.edu/goldenPath/help/bedgraph.html
bigWig:http://genome.ucsc.edu/goldenPath/help/bigWig.html
八、VCF文件(变异记录文件格式)
vcf文件做记录个体或群体突变的文件格式,在生物信息学应用中举足轻重。主流的生物信息分析软件,在处理变异信息时,也基本上需要考虑支持解析或输出vcf格式的文件。本文在介绍vcf文件格式的基本格式的同时,对vcf文件记录的细节进行描述。希望对广大开发者和生物信息学从业人员起到帮助。
1. vcf文件概述
vcf文件格式是变异结果存储的标准格式,一般多用于单核苷酸变异(SNV)或小片段的插入缺失(indels)的结果记录。除此之外,vcf文件也可以存储其他变异形式,比如CNV(拷贝数变异)、SV(结构变异)等,但目前难以形成主流。基因组结构类变异,目前相对较多的依然是bed或bedpe文件,后面会陆续为各位进行介绍。
SNV 是基因组上单个位置的替换。|比如,在参考基因组上记录为 A ,通过检测,某个体由于个体差异或突变,在相同位置变异为C。Indel是指插入或缺失,例如在参考基因组上,某位置为ATCCA,在个体基因组上为ACA(中间位置的TC缺失),则记为A--CA,该位置为deletion。同理若个体基因组存在插入(insertion)情况,与deleltion一样也可以进行记录。insertion和deletion合称为indel。
vcf文件主要有三种模式:
- 第一种为仅有位点信息,即对变异发生的位置和变异本身
- 第二种为个体变异记录的是某个个体或个体组织的突变情况
- 第三种为群体变异检测信息,记录的为突变在群体或家系中发生情况。
这三种类型文件虽然在记录内容上有所差别,但是都遵循vcf的基本规则。下图为vcf文件实例:
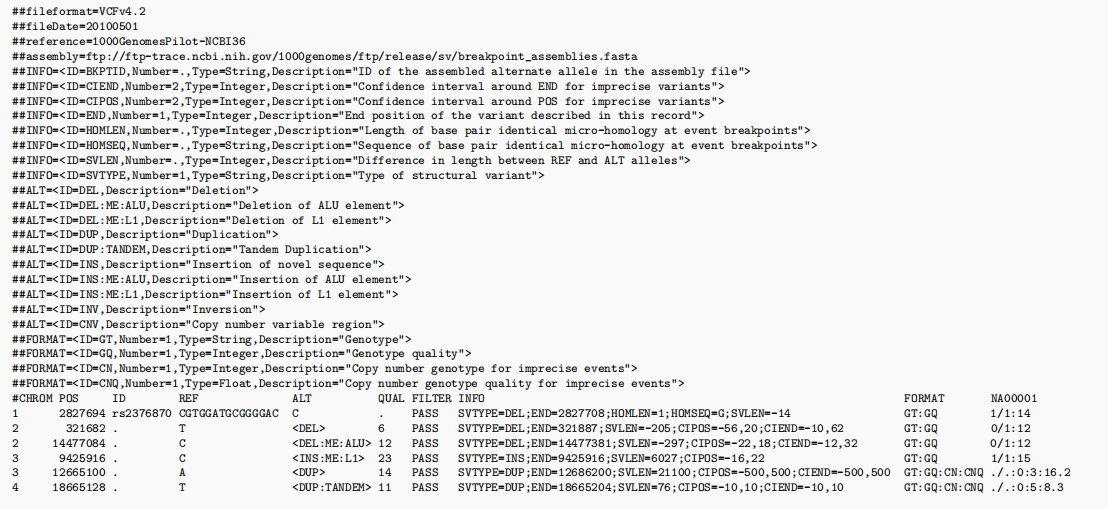
注意: 从上述说明中可以看出,单核苷酸的记录其实相对容易,但是对indel变异而言,由于插入缺失片段的长度不定,其位置并非固定的,因此在样本间的记录难以统一。
2. vcf meta 信息部分
vcf文件头部主要记录文件原信息(meta information),每行以##开始,主要记录内容包括下列内容
| 记录内容 | 说明 |
|---|---|
| 文件格式(fileformat) | 记录文件格式;主要记录本文件所采用的vcf格式版本 |
| 信息区域(INFO)描述 | 该部分记录的是作为INFO区的基本描述,由于在vcf记录中,对INFO内容采用的是缩写,因此在此部分进行会对其含义以及数据类型进行说明。 |
| 过滤方式(FILTER) | 该部分主要记录vcf文件经过生成过程中经过哪些过滤程序处理。记录包括命令行及对应参数 |
| 个体区域格式信息(FORMAT) | 对于每个样本,vcf正文部分会记录多种信息,例如变异对应深度,该部分记录个体信息中每个字段含义及类型 |
| 突变类型(ALT) | 标识出现在正文信息中突变类型,例如,Insertion、Deletion、Duplication 等 |
| 基因组信息(assembly) | 标识对应的基因组版本 |
| Contig信息 | 由于同一物种肯定存在多个基因组版本(如人类目前常用的有hg19和hg38两个版本),因此可能存在染色体名称相同,但内容不同的情况。因此该部分记录主要用于记录每个染色体的相信信息 |
| 样本基因组信息 | vcf理论上支持不同样本比对于不同基因组的,该部分记录样本和基因组的对应关系(通常较少使用) |
| 遗传谱系信息( Pedigree) | 记录样本间的亲缘关系 |
3. vcf 列名称及正文
vcf文件meta信息记录结束之后,下一行就是列名称,列名称以#作为起始,主要包括以下几列:
- CHROM:表示该列为变异所在的染色体位置。
- POS:表示变异起始坐标,该部分需要注意的是对于结构变异,如Indel,其坐标记录方式不唯一。
- ID:表示变异ID,一般常用的有rs编号或vep编号等。用户也可以根据具体问题自行设定编号,空缺时记为“
.”。 - REF:表示位点在参考基因组上的记录。
- ALT:表示位点可能出现的变异情况。
- QUAL:以Phred(即-log10)格式表示变异可靠性。通过不同变异检测软件得到的结果中,该项记录一般无可比性。
- FILTER:表示过滤方式该部分用户也可以自定义,对于不同过滤标准可以进行不同的命名;但是对于通过过滤标准的,统一记为
PASS。需要注意的是,对于一般的vcf处理软件来说,FILER只标记某个变异是否通过过滤条件。而不是删除记录。 - INFO:该部分记录的变异的评价性指标以及注释信息,例如
AC表示的等因突变的数量。该部分在不同的变异检测软件给出的结果也是不同的,大多数基础统计可以通过bcftools软件来进行追加。
上述八列信息是vcf中必然含有的部分,对于有样本信息,无论是样本还是单样本,都会有第9列 FORMAT列及后续样本信息列:
- FORMAT:个体区域格式信息记录样本变异的记录格式。由于vcf文件样本记录中除了变异本身是否发生之外,还会记录针对变异的其他信息,例如对应位点的深度(AD)、基因型质量(GQ)等。FORMAT字段规定了这些信息的基本格式。
- 个体信息: 个体信息是记录某个样本的具体变异情况,其包含的基本信息格式标准需要符合记录的FORMAT格式。特别需要注意的是基因型GT信息,如果是没有经过phased数据,以
/分隔两个等位位点;如果经过phased则以|分隔。其余信息,均会在FORMAT字段有详细说明。
4. vcf记录标准问题
虽然vcf可以记录和表述突变具体情况,但是实践中由于不同变异检测工具对变异判定的区别,会造成对于同一个变异信息存在不同纪录的可能性(尤其是Indels类型记录)。因此,对于vcf文件而言,除了要求记录的信息全面这个最基本的要求之外,还需要做到两个标准。
4.1 简约性(Parsimony)
简约性是指,用尽可能少的核苷酸表示变体,而不会将任何等位基因的长度减少到0。其规定主要针对多核苷酸多样性而言(简称为MNP,指连续多处核苷酸发生突变)。下图列举了4种例子:

这四种颜色(红、绿、橙、蓝)的对于表示同一突变,其实是等效的。但是前三种记录均不是最简方法。因此,只有第四种蓝色的记录方式,符合简约性规范。
4.2 左对齐(Left alignment)
左对齐主要针对Indel类型的变异。其是指,当且仅当不再可能将其位置向左移动,同时保持其所有等位位点的长度不变时,则变异已左对齐。例如下图:

图中给出样本相对对于参考的基因组缺失“CA”碱基的5种记录方式,符合左对齐且简约的,只有最后一种(即紫色)对应的记录方式。
4.3 为什么要遵循上述原则
上述两个原则,其实并不是vcf文件格式规定的,但是相对而言比较重要。尤其对于变异的后期注释、挖掘和使用中,如果记录不满足上述标准,是很难对样本间进行合并或解读。
因此,一般拿到不明来源的vcf文件,比较良好的习惯就是对数据进行标准化处理,再进行其他操作。一般而言,GATK和bcftools都提供了相应的功能,用户可以方便实现。
5. 结构变异
vcf文件设计之处,对于结构变异的检测还不想目前这么百花齐放。因此虽然在vcf格式设计上考虑了结构变异的问题,但是相对而言记录比较粗糙。目前各类检测软件虽然基本上都保留了将结构变异输出成vcf的功能。
在vcf中结构变异以以下形式展现
| REF | ALT | 含义 |
|---|---|---|
| s | t[p[ | 片段p在以从右侧开始的顺序,从t所在位置取代了s |
| s | t]p] | 片段 p 反转后(从左侧),从t所在位置取代s |
| s | ]p]t | 片段p在以从右侧开始的顺序,在t之前的位置取代s |
| s | [p[t | 片段 p 反转后(从左侧),从t之前位置取代s |
例如:

由上面的例子可以看出,采用vcf记录结构变异实在不是一个好主意。相比之下,采用bed文件就更加清晰明了。在后面的内容中会向读者陆续介绍。
6. 小结
vcf作为最通用的变异记录文件格式,其格式的表达内容非常丰富,经过压缩后可以快速查询和定位。同时,基于该格式的应用种类相当多,因此是生物信息学重要的文件格式标准之一。
除了格式要求的明规则之外,使用vcf的时候还应该遵守"潜规则",目前主流变异检测软件对规范化的要求明显有所提高,因此最好在使用之前,进行一系列校正。
最后,vcf文件也支持表达结构变异,但是其人工阅读难度和可解析度其实都不太尽如人意。主流的注释工具更多的支持bed或其他格式的结构突变结果。因此,奉劝大家谨慎使用vcf记录结构变异。
九、GenePred格式(表示基因结构和注释
GenePred是一种用于表示基因结构和注释的表格格式。它是一种比较简单的基因结构描述方式,主要用于基因组注释和基因表达分析。
GenePred格式的每一行描述一个转录本(transcript)或基因(gene),并包含以下列:
name:基因或转录本的名称。chrom:染色体名称。strand:方向,表示基因或转录本的正向(+)或负向(-)。txStart:转录本或基因的起始位置。txEnd:转录本或基因的结束位置。cdsStart:编码序列(CDS)的起始位置。如果基因没有CDS,则为起始位置与结束位置相同。cdsEnd:编码序列(CDS)的结束位置。如果基因没有CDS,则为起始位置与结束位置相同。exonCount:exon的数量。exonStarts:以逗号分隔的exon起始位置列表,相对于转录本起始位置。exonEnds:以逗号分隔的exon结束位置列表,相对于转录本起始位置。
每个转录本或基因可以有多个exon,其起始位置和结束位置根据exonCount、exonStarts和exonEnds进行描述。
GenePred格式是一种简洁而灵活的注释文件格式,经常在基因组注释和基因表达分析中使用。它可以与一些生物信息学工具和软件进行交互,并用于计算基因的长度、外显子数量等信息。
参考:
https://blog.csdn.net/qazplm12_3/article/details/108162714
https://genome.ucsc.edu/FAQ/FAQformat.html#format13
wiggle(WIG):https://genome.ucsc.edu/goldenPath/help/wiggle.html
bedGraph:https://genome.ucsc.edu/goldenPath/help/bedgraph.html
bigWig:http://genome.ucsc.edu/goldenPath/help/bigWig.html